-
5分钟安装免费分子对接工具MGLtools
-
5分钟安装分子动力学模拟软件Gromacs-2020
-
Autodock vina 从安装到分子对接
没在win10用过vina,这次安装一个玩
官网下载MGL tools,处理蛋白和小分子的一个免费工具
http://mgltools.scripps.edu/downloads
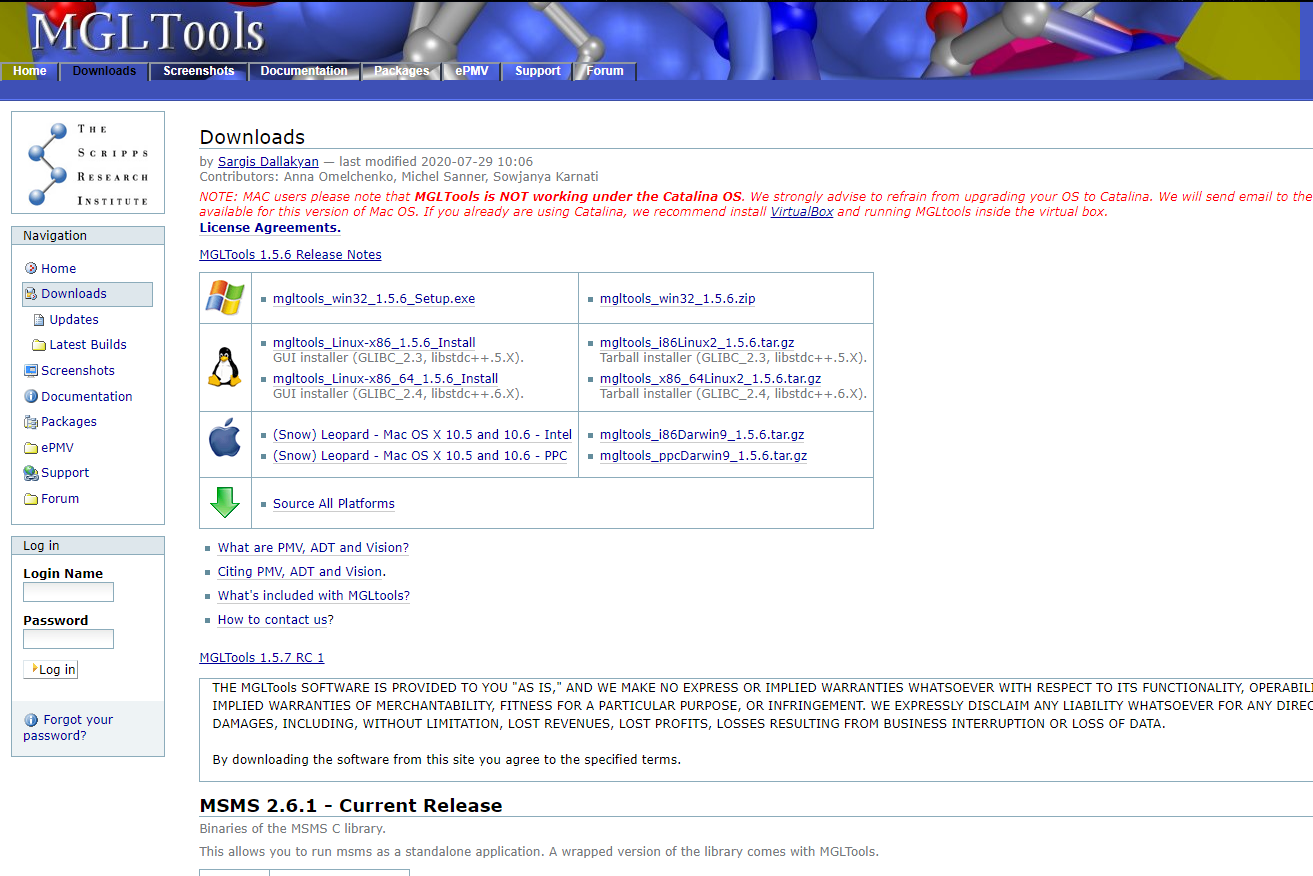
下载win版本,不过只有32位的
安装
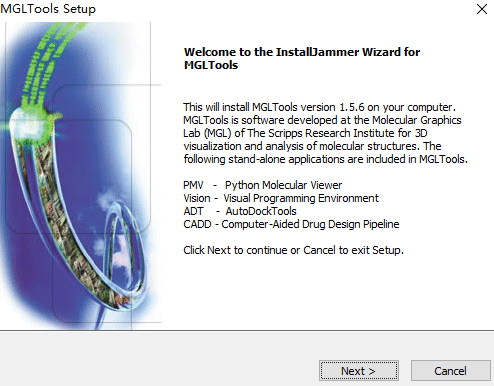
安装的过程中下载vina
也是只有32位的
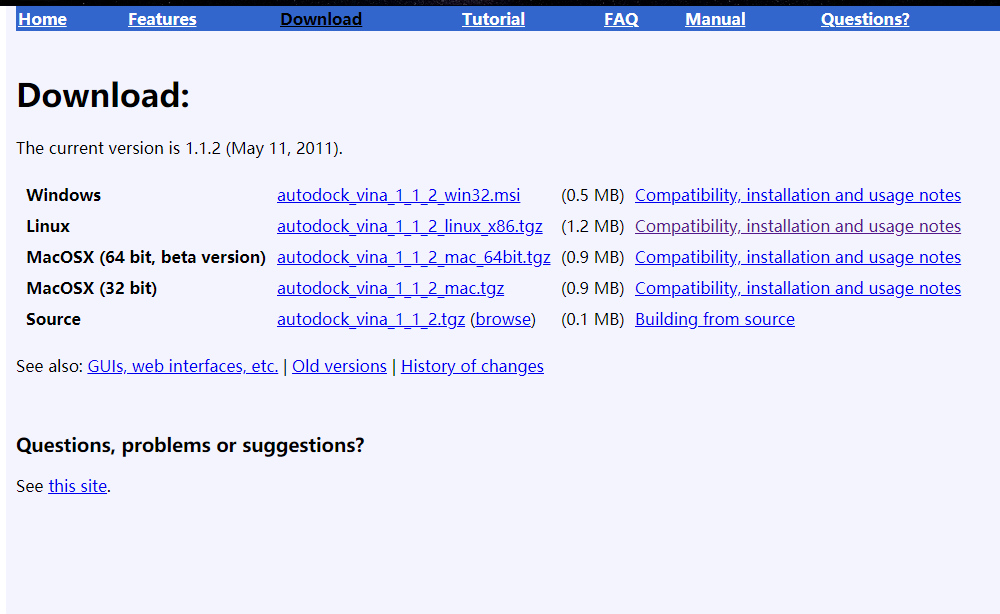
vina直接安装就可以了
MGL tools会同时安装PMV 和CADD

下面软件都准备好了
先用MGL 生成vina需要的pdbqt文件
MGL tools的作用就是生成pdbqt文件
首先打开受体蛋白的pdb
file-read molecule
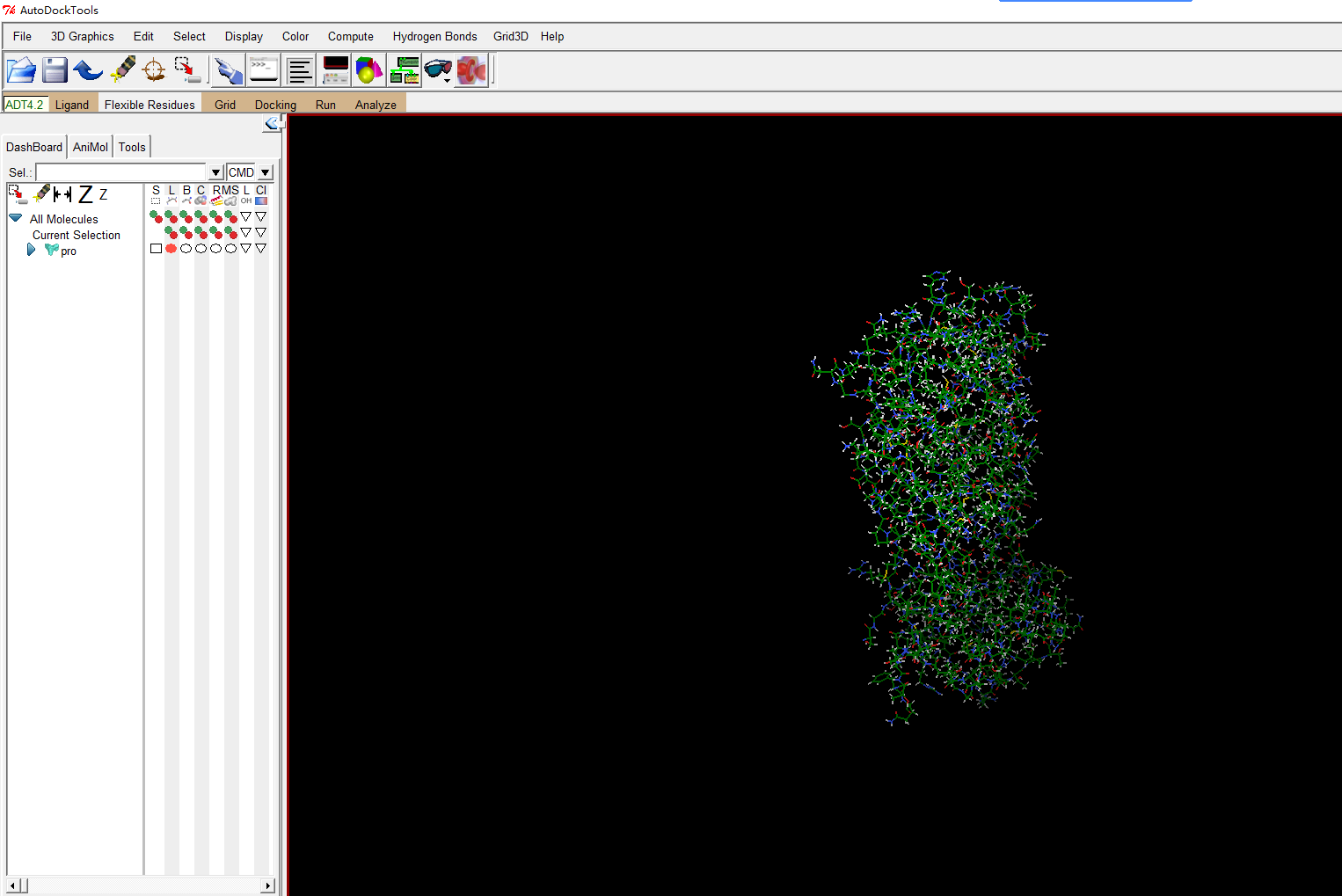
删除水分子和其他配体,常规操作不用解释
然后计算电荷和添加原子类型
Edit–Charges–Compute Gasteiger
Edit–Atoms–Assign AD4 type
就可以导出成pbdqt格式的文件了
然后右键吧蛋白删除掉,导入配体小分子,随便从ZINC下了一个
Ligand-input-open
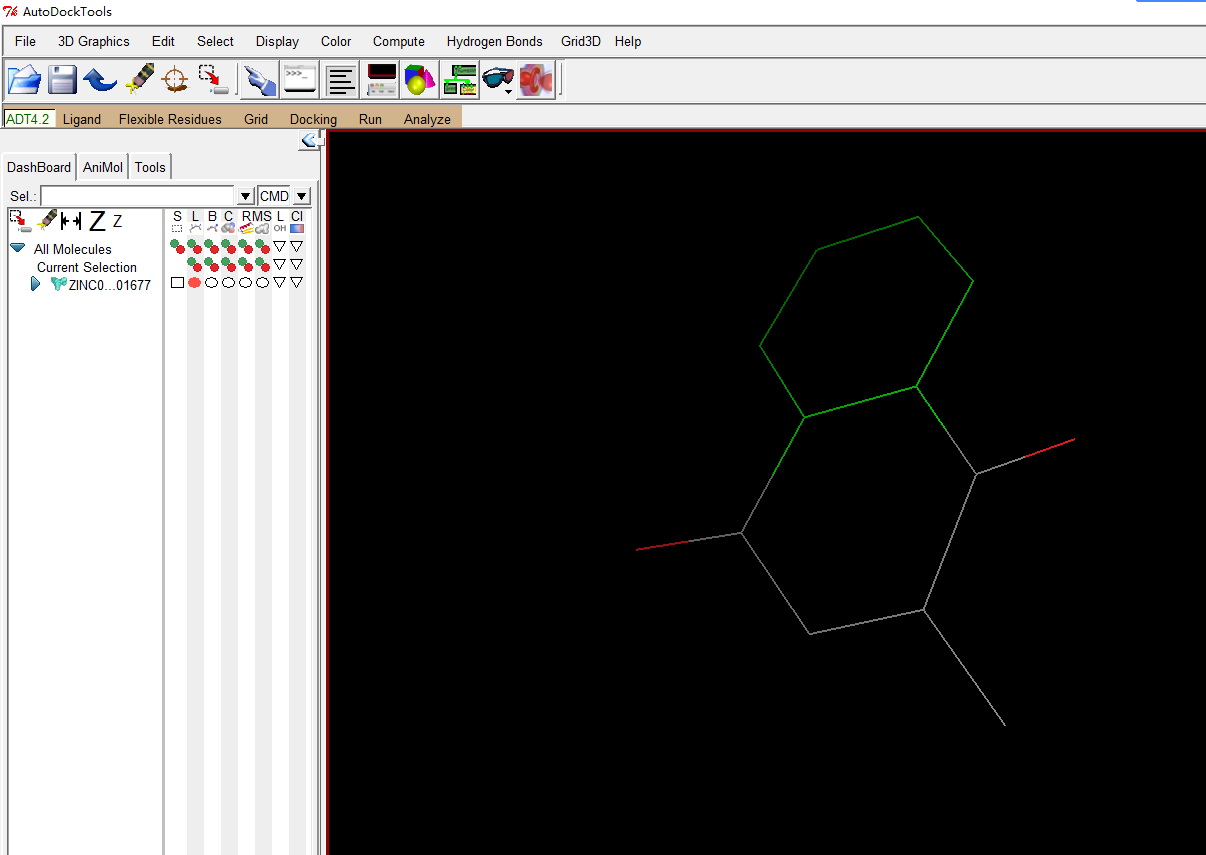
同样的处理方式,加极性氢,计算电荷和添加原子类型
Edit–Charges–Compute Gasteiger
Edit–Atoms–Assign AD4 type
配体因为是半柔性,还需要指定root和可旋转键
Ligand–Torsion Tree–Detect Root
Ligand–Torsion Tree–Choose Torsions
保存
Ligand–Output–Save as PDBQT
和受体放在同一个文件夹
现在开始处理vina,vina需要两个可执行程序,vina_split.exe和vina.exe
刚才我们安装过了vina,所以把这两个程序的目录加入环境变量,或者直接把这两个文件复制到对接的工作目录。
那么现在就有了配体和受体的pdbqt文件,还需要指定对接参数,包括以下
Input:
–receptor arg rigid part of the receptor (PDBQT)
–flex arg flexible side chains, if any (PDBQT)
–ligand arg ligand (PDBQT)
Search space (required):
–center_x arg X coordinate of the center
–center_y arg Y coordinate of the center
–center_z arg Z coordinate of the center
–size_x arg size in the X dimension (Angstroms)
–size_y arg size in the Y dimension (Angstroms)
–size_z arg size in the Z dimension (Angstroms)
Output (optional):
–out arg output models (PDBQT), the default is chosen based on
the ligand file name
–log arg optionally, write log file
Misc (optional):
–cpu arg the number of CPUs to use (the default is to try to
detect the number of CPUs or, failing that, use 1)
–seed arg explicit random seed
–exhaustiveness arg (=8) exhaustiveness of the global search (roughly
proportional to time): 1+
–num_modes arg (=9) maximum number of binding modes to generate
–energy_range arg (=3) maximum energy difference between the best binding
mode and the worst one displayed (kcal/mol)
Configuration file (optional):
–config arg the above options can be put here
Information (optional):
–help display usage summary
–help_advanced display usage summary with advanced options
–version display program version
修改之后写如conf文件中,其中energy_range是和最佳模型的能量值相差的最大值,单位kcal/mol,一般-7kcal/mol才有意义,所以可以设置为2-4,其他的就不需要动了
打开终端到工作目录,开始计算
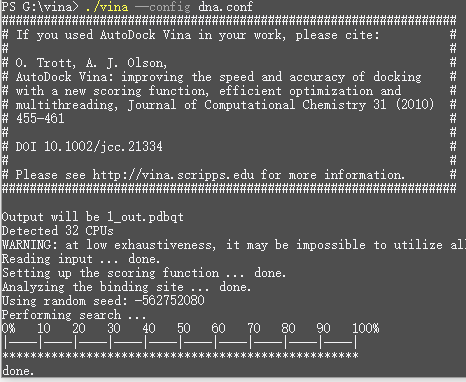
结果直接用pymol打开就可以了
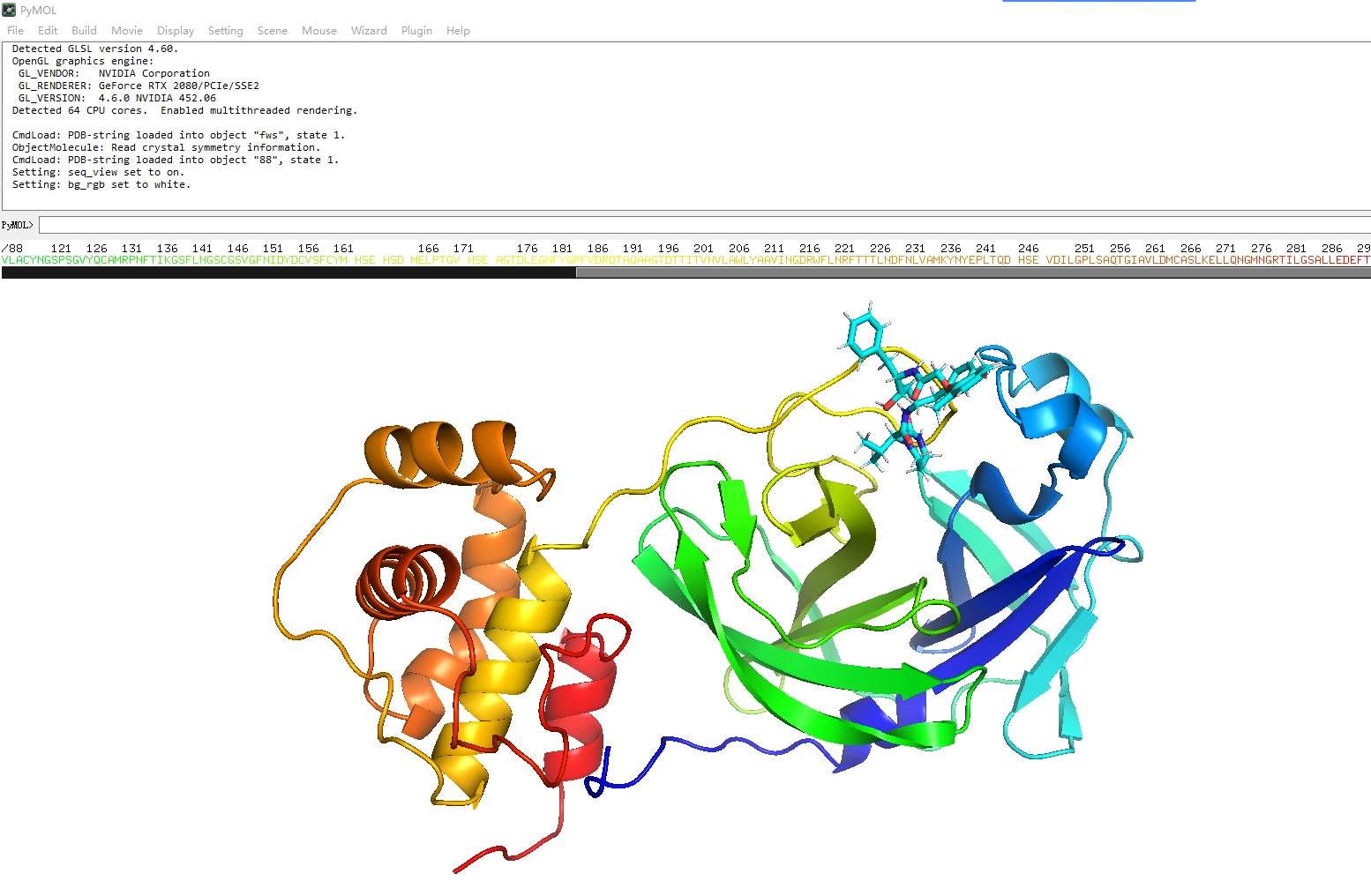
-
Western Blot实验全面总结-1 上样缓冲液配制
本来想从头到尾按照实验顺序写一个总结,结果今天正好做实验的时候SDS-PAGE的loading_buffer没了,临时改造了琼脂糖凝胶的loading_buffer,就顺便先从loading_buffer开始写了。这个东西也是大多数人容易忽略的细节。
loading_buffer的作用是让蛋白样品变成可以用于电泳的状态,主要功能包括:
1,平衡蛋白电荷。由于电泳时电压相同,会造成蛋白会根据自身电荷水平出现电泳速度差异,而不是分子量。这样就无法通过分子量区分不同的蛋白条带,所以缓冲液第一个成分就是SDS,平衡电荷。
2,减少上样误差。误差主要存在于上样的时候从孔中溢出, 改变的方法就是用甘油把样品的密度加大,让样品沉降到孔的底部,所以第二个成分是甘油。
3,保持蛋白变性。电泳依赖于蛋白变性,完整的蛋白是一坨三级结构,要变成线性才能通过凝胶的微孔,变性通过煮沸就可以,但是冷却后蛋白会自发恢复三级结构,所以需要加还原剂保护巯基。这个部分的成分就是DTT或者β-巯基乙醇。
4,标记颜色。上样的时候需要知道自己的样品是不是进入了孔中,跑电泳一半也需要大概看一下溴酚蓝的条带,不过这不是必选项,不加也可以。
那么好了,直接配制
首先要知道,上样缓冲液有AB两个组分,需要使用的时候混合,就是还原剂需要现用现配,所以长期保存的A液就是SDS/甘油/溴酚蓝。B液就是β-巯基乙醇。
5X SDS-PAGE load_buffer 配方(15ml+5ml)
A液:
甘油 5 mlSDS 1g
溴酚蓝(2%) 2.5 ml
Tris-HCL(1M,pH=6.8) 定容至10ml
B液:
β-巯基乙醇 5ml用前加入3:1=A液:B液